We put the example sequencing reads in the home directory, you can check this fastq file by:
% head /home/reads_1.fq
% head /home/reads_2.fq
Question: How many reads are in the reads_1.fq file? How do you count the number using linux command?
Stay in the directory, which contains the lambda_virus index files you created in the previous step. Next, run:
% bowtie2 -x lambda_virus -U /home/reads_1.fq -S eg1.sam
This runs the Bowtie 2 aligner, which aligns a set of unpaired reads to the Lambda phage reference genome using the index generated in the previous step. The alignment results in SAM format are written to the file eg1.sam, and a short alignment summary is written to the console. (Actually, the summary is written to the "standard error" or "stderr" filehandle, which is typically printed to the console.)
To see the first few lines of the SAM output, run:
% head eg1.sam
Bowtie 2 outputs alignments in SAM format, enabling interoperation with a large number of other tools (e.g. SAMtools, GATK) that use SAM. For details about the format, see the SAM Format Specification
Read the sam format specification, and try to find out the mapping position of sequencing read "r6".
To align paired-end reads, stay in the same directory and run:
% bowtie2 -x lambda_virus -1 /home/reads_1.fq -2 /home/reads_2.fq -S eg2.sam
To see the first few lines of the SAM output, run:
% head eg2.sam
The Integrative Genomics Viewer (IGV) is a high-performance visualization tool for interactive exploration of large, integrated genomic datasets. It supports a wide variety of data types, including array-based and next-generation sequence data, and genomic annotations.
Download the reference genome lambda_virus.fa and your alignment result SAM file: eg1.sam into your local machine.
Download the IGV from Broad Institute: IGV download .
After installation of IGV, run the application
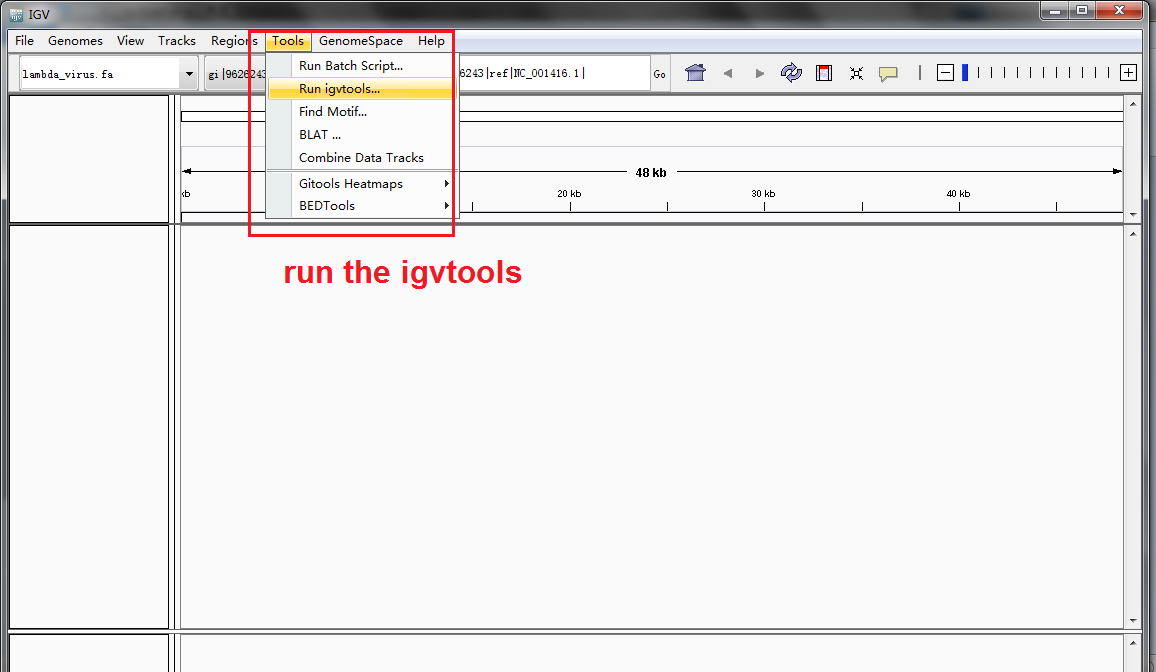
Run igvtools for SAM sorting and indexing
Sort the SAM file from your alignment 
Build up index for the sorted SAM file 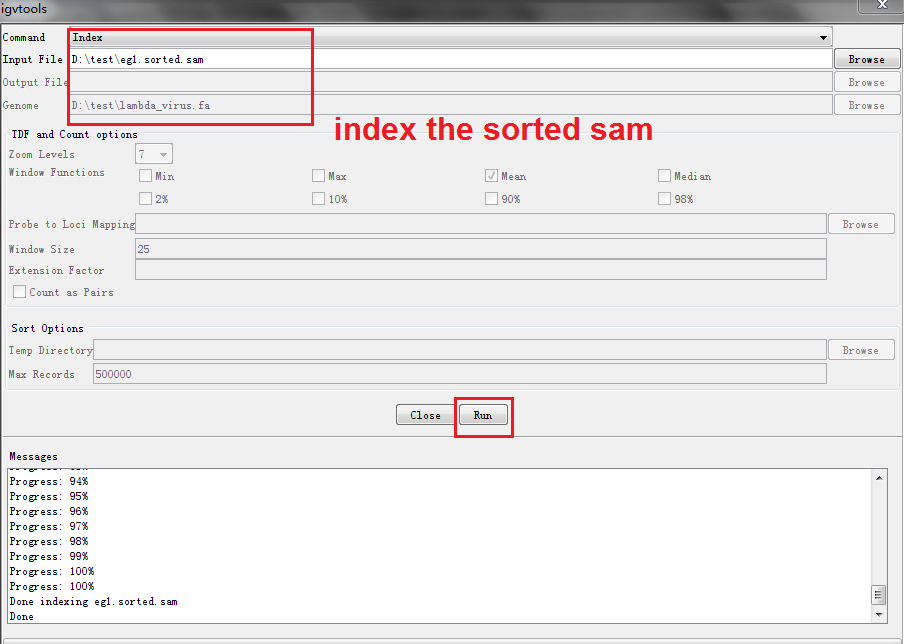
Load our reference genome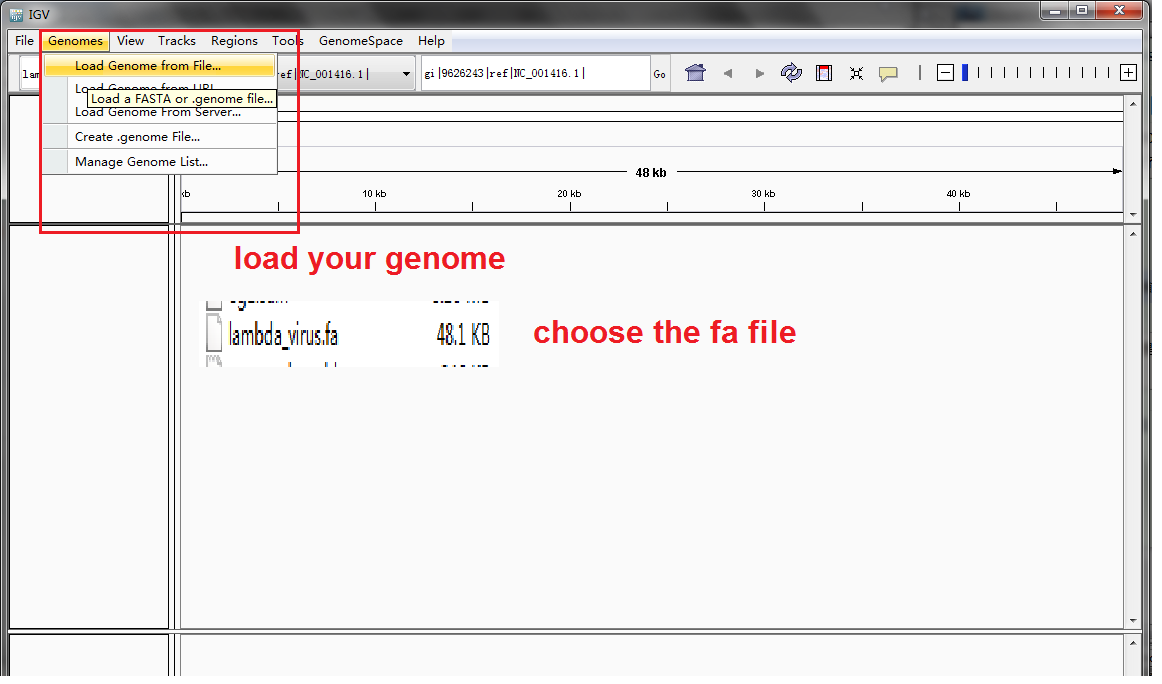
Visualize and browse the mapping result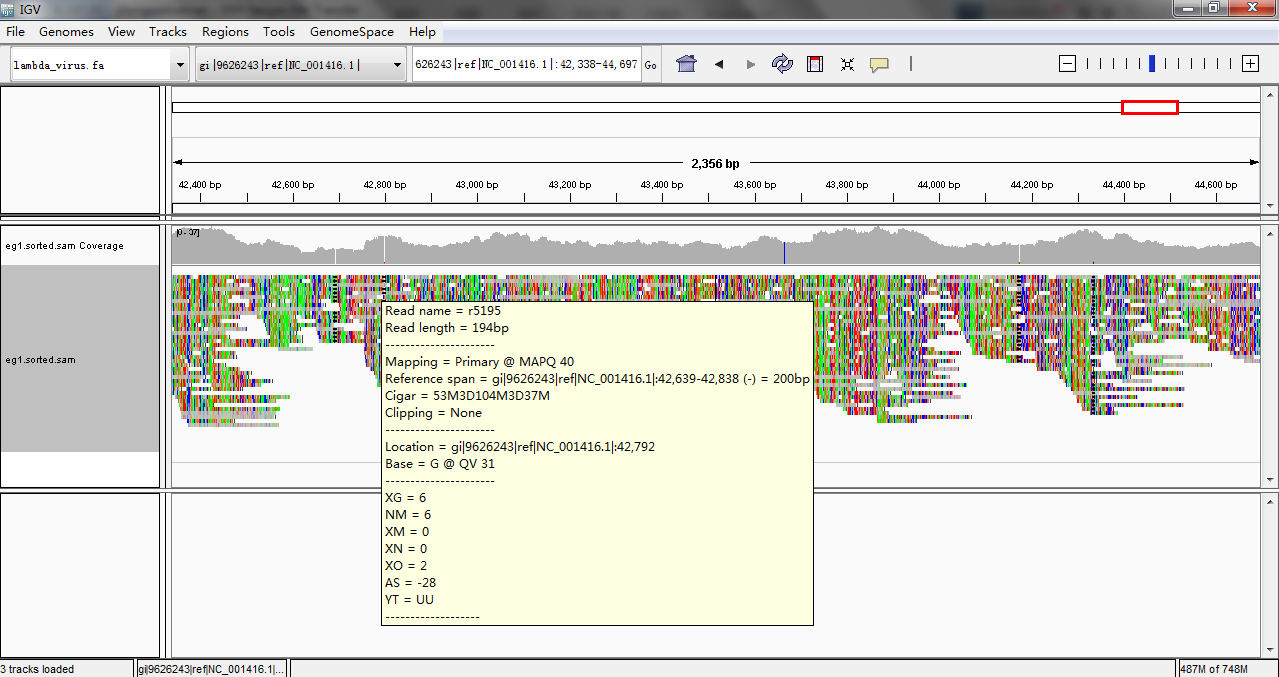
bowtie2 |
align the sequencing reads |
bowtie2-inspect |
extract the summary information of the indexed reference genome |
tongyinbio@hku.hk bbru@hku.hk 13th-Feb 2017